Ectopic activation of suicide inhibitors
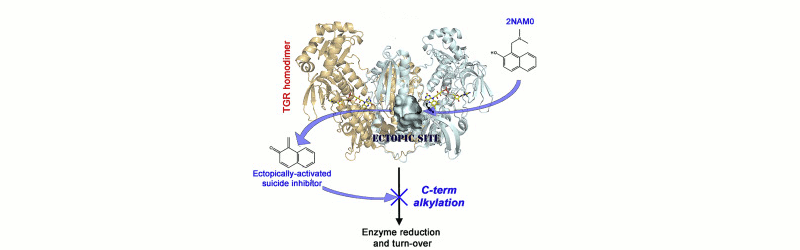
Thioredoxin reductases (TR), and the related Thioredoxin Glutathione Reductases (TGR), are attractive targets for the therapy of chronic inflammatory diseases, cancer, and parasitoses. With rare exceptions, these enzymes are selenoproteins and can be inhibited irreversibly by drugs targeting their redox-active Se-Cys residue.
Several drugs target TRs: e.g auranofin, a gold-containing compound used in the treatment of rheumatoid arthritis, which has also anticancer and antiparasitic activity against malaria and schistosomiasis.
The high reactivity and low pKa of the selenol group, which make Se-Cys highly reactive, impair the specificity of irreversible inhibitors; thus irreversible inhibitors of TRs and TGRs may have numerous side effects.
A recent paper by Silvestri et al. describes an elegant strategy to circumvent this problem. The authors studied a family of molecules that may be activated by the TGR from the human parasite Schistosoma mansoni and converted to irreversible inhibitors. Activation occurs at a site of the protein unrelated to the catalytic site and quite removed from the target Se-Cys residue. The site of activation is close to the binding site of NADPH, the reducing substrate of TRs and TGRs, and is not present (or at list has not the same structure) in human TRs. The precursor molecules are 2-naphtholmethylamino derivatives that are ectopically converted by TGR to quinone methides; these, in turn, alkylate the Se-Cys residue of the enzyme causing its irreversible inactivation. The strategy outlined in the above study may not be generally applicable: after all every enzyme has a substrate binding site, but few enzymes may have ectopic sites suitable for the activation of suicide inhibitors; yet, it may be well worth pursuing for those enzymes that are attractive drug targets and whose known inhibitors lack species-specificity.




Join the FEBS Network today
Joining the FEBS Network’s molecular life sciences community enables you to access special content on the site, present your profile, 'follow' contributors, 'comment' on and 'like' content, post your own content, and set up a tailored email digest for updates.