AlphaFold protein structure predictions - a step change for biology

This post was written by Sameer Velankar, Gerard Kleywegt and Alex Bateman, all from EMBL’s European Bioinformatics Institute (EMBL-EBI). Watch the online talk about the AlphaFold database that Sameer Velankar and Gerard Kleywegt gave to the FEBS Junior Section on 26 October 2021.
The protein folding problem was first introduced in 1968 and referred to the challenge to predict the 3D structure of a protein based solely on its sequence of amino acids. Since then, it has remained one of the biggest unsolved mysteries in biology, despite numerous research efforts. This all changed in November 2020, when the AlphaFold artificial intelligence system developed by DeepMind seemed to have cracked the problem.
Less than a year later, more than 350,000 AlphaFold protein structure predictions have been made freely and openly available for anyone to access through the AlphaFold Database (AlphaFold DB), co-developed by DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI). This number is set to increase to over 100 million structures in the near future.
The power of open data
Since the 1970s, the structural biology community has archived experimental protein structures in the Protein Data Bank (PDB), a freely available global resource that now contains over 180,000 structures. The AlphaFold system builds on this huge body of experimental information and generates its predictions by analysing the relationship between these known protein structures and huge amounts of protein-sequence data. This protein-sequence information has also been generated by scientists all over the world, mainly through genome sequencing, and is made available through public resources, such as UniProt and MGnify hosted at EMBL-EBI. As a result, AlphaFold is able to produce near experimental-quality structure predictions for a wide range of proteins. AlphaFold is the first AI method to achieve such good results, as demonstrated in the CASP14 conference.
DeepMind and EMBL-EBI have now partnered to make hundreds of thousands (and eventually many millions) of AlphaFold structure predictions freely available through AlphaFold DB. The initial release of the database includes structure predictions for 98.5% of the proteins in the human proteome. By contrast, only 11% of human proteins have had their structure determined experimentally.
In addition to structure predictions for almost the entire human proteome, AlphaFold DB also includes protein structure predictions for 20 other species of significant biological or medical interest. In the coming months, the resource will grow to cover a large proportion of all catalogued proteins (a reference sequence for each cluster that groups sequences with up to 90% sequence identity; i.e. UniRef90).
This means that for almost every known sequence in UniProt there will be either an experimentally determined structure in the PDB, or a predicted structure in AlphaFold DB, or a template structure in the PDB or AlphaFold DB with >90% sequence identity to the protein of interest. The source code of AlphaFold has been made open as well, so predictions for newly discovered and non-natural (designed) sequences can be generated by anybody who wants to.
Being aware of the limitations
Although the availability of predicted 3D models for the known “protein universe” is an exciting prospect with huge impact, there are certain limitations to the AlphaFold method and database that researchers need to be aware of. The most important caveat is that AlphaFold structures, although high quality, are still predictions and most of them have not yet been validated experimentally. Below are some of the limitations of the method:
- The AlphaFold method is not designed to predict structures for complexes of protein with other proteins, nucleic acids or small molecule ligands. Many proteins function as complexes, hence the predicted structures available in the database may not necessarily provide insights into the function of the protein.
- AlphaFold models do not include any non-protein components such as cofactors, metals, ligands including drug-like molecules, ions, carbohydrates and other post-translational modifications.
- Each structure in the database represents a possible conformation of the protein of interest.
- Proteins are dynamic systems and adopt multiple conformations depending on their environment or state within a functional cycle. Availability of the predicted structures may inspire and support experimental studies of conformational dynamics of proteins, which are crucial for understanding biological function.
- AlphaFold can produce regions that are predicted with low-confidence and adopt an extended conformation. These regions often correspond to regions that are intrinsically disordered or unstructured.
- Some mutations can destabilise a protein so that it will not fold up in vivo. AlphaFold is not trained to recognise this and will provide a folded structure in such cases.
The predicted structures may (or may not) lead directly to mechanistic or functional insights regarding the protein. However, the models themselves not only correspond to over 350,000 structural hypotheses, they may also lead to many other hypotheses about domain structure, function, specificity, interactions, binding partners, roles of mutations, etc. for equally many proteins, and all these hypotheses can be tested experimentally.
Opening up new research avenues
The protein-structure predictions in AlphaFold DB will have an immediate impact on molecular structural biology research, and in the long term, a potentially significant scientific, medical and economic impact.
AlphaFold DB could also open up the development of research areas that were previously impossible, impractical or limited by the relatively restricted amounts of 3D structural information available. PDB contains over 180,000 entries which cover ~55,000 unique proteins. This is a tiny fraction of the number of known protein sequences, which is estimated at 220 million sequences in UniProt, or even more in metagenomics databases such as MGnify (>600 million). Imagine what discoveries the millions of protein structure predictions set to be made available in AlphaFold DB can help to unlock.
The availability of predicted 3D models on a large scale is likely to accelerate structure studies by kick-starting experimental structure determination. AlphaFold can also help to elucidate structures for which experimental data was collected years ago, but the phasing problem could not be solved previously. In the immediate aftermath of the announcement of AlphaFold DB there have been a considerable number of anecdotal reports of such cases.
The availability of 3D models for almost every protein is also likely to shift the focus of structural biologists to different challenges, such as predicting the structure of protein complexes and assemblies, and predicting how drugs and other small molecules interact with proteins. After all, proteins exist in dynamic systems and not in isolation.
In the field of drug discovery, the use of 3D models can provide an understanding of why a certain drug compound is an inhibitor while a related compound is not, or why certain proteins are “druggable” while others are not. Researchers can also use virtual screening techniques to find new uses for existing drugs.
Crucially, deep-learning technology may become a mainstay of future developments to understand how proteins work. One such challenge is that of studying intrinsically disordered and mobile regions in proteins, which are functionally important and may adopt a well-defined structure in the appropriate context, for example when interacting with a partner protein. About a third of the human proteome is predicted to contain intrinsically disordered regions and insights provided by the advances in prediction methods will advance their study.
There are too many potential research avenues to tackle in this article, but if you would like to find out more, you can read a more detailed analysis here. Or why not take a dive into the AlphaFold DB to see what wonders you can uncover?
The possibilities may be endless, and one thing is certain: AlphaFold DB represents the beginning of a very exciting time in biology and a reinvigoration of protein science that will drive innovation in the coming decades.
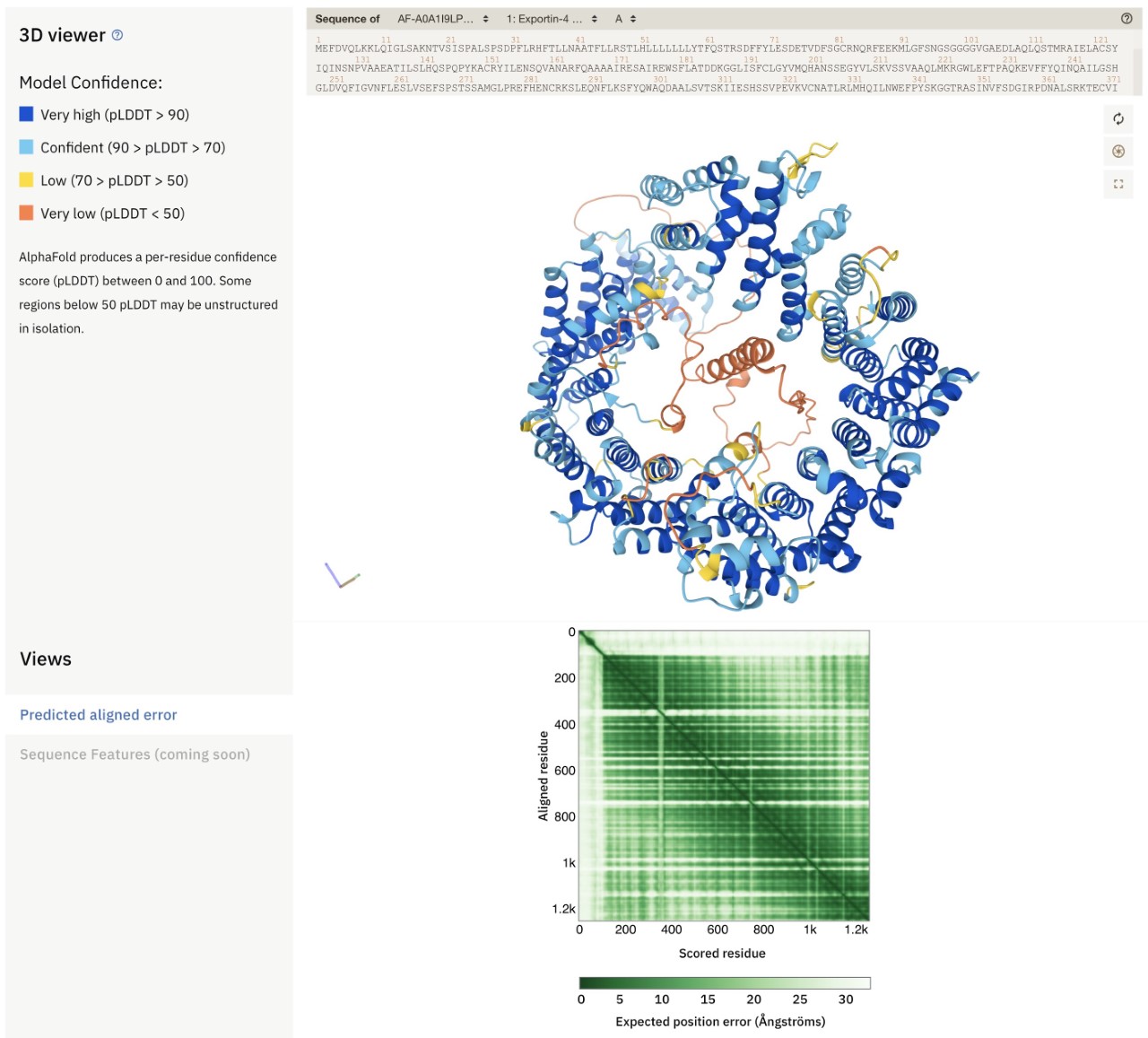
Example of an AlphaFold prediction from AlphaFold DBThe image below is an example of an AlphaFold prediction from AlphaFold DB. The structure is for the Arabidopsis exportin-4 protein (https://alphafold.ebi.ac.uk/entry/A0A1I9LPW2), which is involved in protein export from the nucleus. This protein is predicted to adopt a donut-like shape, with an N-terminal helix inside the hole of the donut. This location of this helix might tempt one to suggest a functional role (acting like a “cork”?), but it is important to assess the predicted quality and reliability of the model before making any such inferences. AlphaFold DB provides two useful visual cues to help biologists with this task. First, the 3D model (top) is coloured by the level of confidence AlphaFold has in its own structure prediction for each residue. Cyan and dark blue regions are predicted with (high) confidence, but for yellow and in particular orange regions the predictions are not generally reliable. The N-terminal helix inside the hole in the donut has very low confidence (as indicated by its orange colour) and hence no inferences should be made from its predicted structure (it may not even be a helix at all). Second, the square plot with green shading (bottom; “Predicted Aligned Error”) provides information about the reliability of the relative positioning and orientation of different parts of a model. In this case the entire N-terminal 100-or-so residues have no green signal with the other residues of the protein. This suggests that any interactions of the N-terminal region and the rest of the protein that may be suggested by the 3D model are likely to be artifactual and not credible. Thus, even if the helix had been predicted with high confidence, its location and orientation inside the donut is not reliable. This example shows the importance for users of AlphaFold DB models to learn how to interpret the models and their reliability so as to avoid over-interpretation. Furthermore, any hypotheses generated from these models should be tested experimentally.
|
This post was written by Sameer Velankar, Gerard Kleywegt and Alex Bateman, all from EMBL’s European Bioinformatics Institute (EMBL-EBI).
Top image by DeepMind.





Join the FEBS Network today
Joining the FEBS Network’s molecular life sciences community enables you to access special content on the site, present your profile, 'follow' contributors, 'comment' on and 'like' content, post your own content, and set up a tailored email digest for updates.
If you are a student or early career researcher interested in this topic, listen to the online talk that Gerard Kleywegt and Sameer Velankar will give to the FEBS Junior Sections on 26 October 2021, 19:00 (CEST). For more information visit:
https://network.febs.org/posts/room-for-junior-sections-of-febs-societies