Hydrogenotrophic methanogens are strictly anaerobic microorganisms that thrive at the thermodynamic limit of Life by oxidizing H2 and reducing CO2 into methane (CH4). They are one of the main contributors to global CH4 emissions, the second most impactful greenhouse gas, but also a valuable biofuel to heat our houses. Therefore, understanding these ancient archaea (more than 3.4 billion years old) and their metabolism is of particular interest in times of climate change. Methanogens live worldwide in environmental niches without oxygen, such as the deeper layers of marine sediments, hydrothermal vents or as part of the gut microbiome, even in human ones. Those environments are rich in sulphides (S2-), which methanogens assimilate as a source of sulphur for their growth and replication. However, sulphite (SO32-), an oxidized form of sulphur, is a poison for them as it inhibits the enzyme generating CH4 and therefore collapses their central energy metabolism.1
The F420-dependent sulphite reductase (Fsr)
Despite its toxicity, certain marine methanogens from the order Methanococcales have been shown to not only tolerate SO32- but even grow on it as a sole source of sulphur.2,3 In other words, they turn poison into food. In their natural environments, they are occasionally exposed to SO32- when oxygenated seawater enters the sediment or vent plumes. Partial oxidation of S2- results in the formation of SO32-, which the methanogens need to detoxify immediately.4 This is possible due to the F420-dependent sulphite reductase Group I or, in short, Fsr – a single enzyme that harbours two catalytic units. One reason why Fsr is very efficient at detoxifying SO32- is because it uses the reduced F420 cofactor as a source for electrons.
F420 is the molecule that gives a bright blue fluorescence to methanogens under fluorescent microscopy (see figure) because of its natural abundance.5 Once F420 is oxidized, it can quickly be recycled by the reducing power of H2 oxidation.6 Furthermore, the overall reaction is highly exergonic, which promotes a fast rate of SO32- reduction:
HSO3- + 3F420H2 → HS- + 3H2O + 3F420 ∆G0′ = -135 kJ mol-1

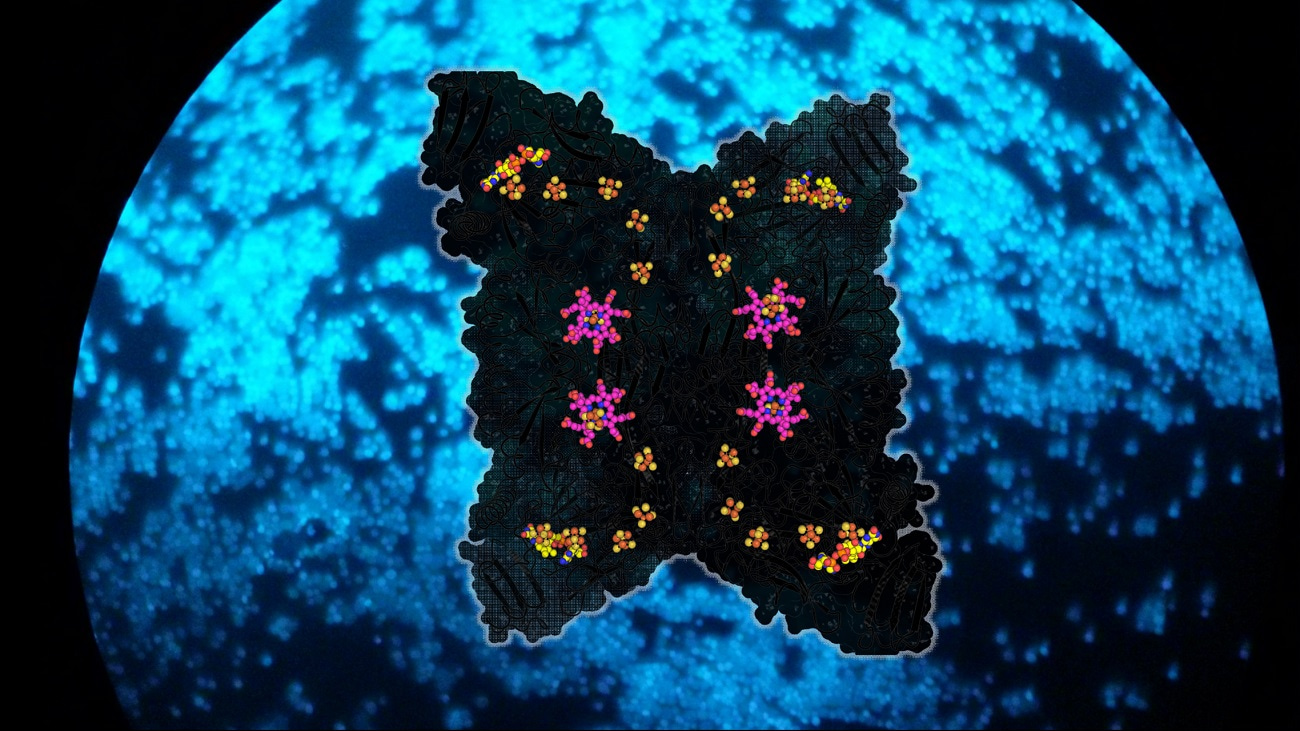
The crystal structures of Fsr from Methanothermococcus thermolithotrophicus and Methanocaldococcus jannaschii elucidated the detailed reaction mechanism of the enzyme. One-half of Fsr, the F420H2-oxidase, transfers the electrons from the F420H2 to its flavin (FAD). From here, the electrons are passed via a six-[4Fe–4S] cluster relay to its other half, a sulphite reductase harbouring a siroheme. The SO32- anion is attracted to the active site of the sulphite reductase by a positively charged cavity, followed by a tight interaction between the SO32- and the siroheme, which carries out the six-electron reduction reaction to generate S2-. The chain of [4Fe-4S] clusters is essential to facilitate the electron flow, and it allows a tight electronic coupling between F420H2 oxidation and SO32- reduction, as attested by electron paramagnetic resonance experiments.
A primitive sulphite reductase
Since Fsr has been isolated from (hyper)thermophilic Methanococcales, it has been shown to have a high-temperature tolerance (>85 °C)2 and it enables the methanogens to survive even high SO32- concentrations (up to 40 mM for Methanothermococcus thermolithotrophicus). Besides being of interest to chemists because of its efficiency and robustness, Fsr's sulphite reductase domain might provide new evolutionary insights. Sulphite reductases are considered to be ancient enzymes that have shaped our Earth for at least 3.47 billion years.7 While dissimilatory sulphite reductases (dSirs, here, referred to as DsrAB) are used by microorganisms to conserve energy, assimilatory sulphite reductases (aSirs) are needed to generate S2- for biomass production (i.e., for amino acids like cysteine, methionine, or cofactors, etc.). Based on structural and phylogenetic studies, it has been proposed that aSirs and dSirs originated from a common ancestor. This progenitor was most likely much simpler than the modern sulphite reductases, which arose through the duplication of the sulphite reductase domain. The crystal structures of Fsr revealed that it contains an interesting mix of features from both assimilatory and dissimilatory sulphite reductases. Its overall architecture resembles a dSir, but the molecular mechanism behind SO32- reduction to S2- is identical to assimilatory ones. Furthermore, Fsr does not contain certain extensions, as reported for aSirs and dSirs, and is the most compact and “primitive” sulphite reductase that has been structurally characterised so far. Because of its “primitive organisation”, Fsr would provide a plausible picture of an ancient sulphite reductase prototype. However, its evolution is still debatable, as the origin of Fsr remains uncertain.
Besides Fsr Group I, the closely related Fsr Group II, which is present in some genomes of anaerobic methane oxidisers (ANMEs) and Methanosarcinales, seems to serve a different function. Fsr Group II, recombinantly produced in a methanogen, did not reduce SO32- but performed nitrite reduction, albeit at a low rate.8
Cultivating hydrogenotrophic methanogens can be challenging as they require a strictly anaerobic atmosphere, a supply of H2 and CO2, and S2- as a sulphur source. However, they are excellent gas bio-converters used in biotechnology. Using SO32- in bioreactors will circumvent the need to use hydrogen sulphide as a sulphur substrate, which is corrosive, explosive and highly toxic for humans. This opens opportunities for safer biotechnological applications and simplifies the studies of these important microorganisms. An optimal solution would be to find a methanogen that reduces sulphate, which is cheap, abundant, and a completely safe sulphur source. One methanogen has already been reported to grow on SO42- – M. thermolithotrophicus.9 Most likely, Fsr orchestrates the last reaction of this sulphate reduction pathway because one of its intermediates would be sulphite. Furthermore, we showed that Fsr from M. thermolithotrophicus reduces nitrite at the same rate as sulphite. Nitrite is another poison that could damage the CH4-generating machinery. However, the physiological contribution of Fsr in this process needs further investigation.
References:
- Balderston, W. L. & Payne, W. J. Inhibition of methanogenesis in salt marsh sediments and whole-cell suspensions of methanogenic bacteria by nitrogen oxides. Science (New York, N.Y.) 32, 264-269, doi:doi:10.1128/aem.32.2.264-269.1976 (1976).
- Johnson, E. F. & Mukhopadhyay, B. A new type of sulfite reductase, a novel coenzyme F420-dependent enzyme, from the methanarchaeon Methanocaldococcus jannaschii. J Biol Chem 280, 38776-38786, doi:10.1074/jbc.M503492200 (2005).
- Jespersen, M., Pierik, A. J. & Wagner, T. Structures of the sulfite detoxifying F420-dependent enzyme from Methanococcales. Nature Chemical Biology, doi:10.1038/s41589-022-01232-y (2023).
- Jannasch, H. W. & Mottl, M. J. Geomicrobiology of deep-sea hydrothermal vents. Science (New York, N.Y.) 229, 717-725, doi:doi:10.1126/science.229.4715.717 (1985).
- DiMarco, A. A., Bobik, T. A. & Wolfe, R. S. J. A. r. o. b. Unusual coenzymes of methanogenesis. 59, 355-394 (1990).
- Vitt, S. et al. The F420-reducing [NiFe]-hydrogenase complex from Methanothermobacter marburgensis, the first X-ray structure of a group 3 family member. J Mol Biol 426, 2813-2826, doi:10.1016/j.jmb.2014.05.024 (2014).
- Colman, D. R. et al. Structural evolution of the ancient enzyme, dissimilatory sulfite reductase. Proteins 90, 1331-1345, doi:10.1002/prot.26315 (2022).
- Heryakusuma, C. et al. A reduced F420-dependent nitrite reductase in an anaerobic methanotrophic archaeon. Journal of bacteriology 204, e0007822, doi:10.1128/jb.00078-22 (2022).
- Daniels, L., Belay, N. & Rajagopal, B. S. Assimilatory reduction of sulfate and sulfite by methanogenic bacteria. Appl Environ Microbiol 51, 703-709, doi:10.1128/AEM.51.4.703-709.1986 (1986).
Top image provided by the author.





Join the FEBS Network today
Joining the FEBS Network’s molecular life sciences community enables you to access special content on the site, present your profile, 'follow' contributors, 'comment' on and 'like' content, post your own content, and set up a tailored email digest for updates.