
Molecular Oncology bridges basic, translational and clinical research to promote advancement in the fields of cancer diagnostics, treatment and care. With the aim of promoting scientific advancements, we have compiled a summary of select content published in our journal in 2022 that covers different aspects of cancer research
1) Developments in basic research are a necessary step for the effective formulation of targeted cancer therapies and as witnessed from articles published last year, particular attention has been paid to identifying underlying factors contributing to carcinogenesis as well as revealing metabolic networks and immune components in tumors:
Lindén et al employed chromatin immunoprecipitation sequencing and proteomic analysis to elucidate that FET oncoproteins, SWI/SNF components and BRD4 co-localize on chromatin and interact with Mediator and RNA Polymerase II, providing a possible molecular mechanism for FET-fusion-induced oncogenic transcriptional profiles.
Prieto-Garcia et al used pharmacological inhibition of USP28 to report that the proteome of transformed cells is reset towards a ‘premalignant’ state and the specific inhibitor could show synergy with clinically approved compounds.
Ruiz-Rodado et al reported the dependence of glioma cells on exogenous cysteine/cystine by exploring nutritional deprivation in a mouse model of glioma and highlighted a time window where cysteine deprivation can be exploited for additional therapeutic strategies.
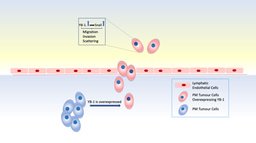
Wilkie et al took advantage of an immune-competent orthotopic breast cancer mouse model to identify breast tumor microbiota was mainly enriched with Gram-negative bacteria, serving as a primary source of lipopolysaccharide (LPS).
García-Gómez et al showed that TGFβ induces NOX4 expression and increases ROS production in a NOX4-dependent manner in several patient-derived glioblastoma stem cells and that is necessary for the expression of stem cell markers and for NRF2, controlling the cell’s antioxidant and metabolic responses.
Rye et al mapped the phenotype and distribution of immune cells in non-metastatic and metastatic sentinel lymph nodes and in metastatic axillary lymph nodes from breast cancer patients to enable the assessment of novel immunotherapy treatments for metastatic breast cancer.
Charpentier et al argued that inducing stress via thapsigargin could boost tumour antigen detection. They identified that drug-induced stress increased translation of a lncRNA-encoded peptide, MELOE-1, which is recognised by a MELOE-1-specific T-cell clone in melanoma cells.
2) Integrating data from patient screening has allowed for the definition of specific gene expression, DNA methylation and metabolic profiles which have been associated with classifying patient risk, prognosis and survival. Such patient characterisation provides an avenue for predicting personalised medicine strategies before treatment.
Papaiz et al combined methylome and transcriptome data to discover the relationship between promoter and/or gene body DNA methylation alterations and gene expression in early, intermediate, and late stages of melanoma progression. Integrating gene expression and DNA methylation profiles from a murine model of melanoma progression revealed potential prognostic markers.
Łysiak et al analysed androgen receptor (AR) genetic alterations in glioblastoma across males and females. They described high AR gene expression in GBM exhibits sex-dependent effects on patient survival with high AR expression correlating with shorter overall survival in females and longer in males.
Classon et al used publicly available sequencing data to examine human leukocyte antigen (HLA) alleles, antiviral T-cell receptors and prostate cancer disease recurrence after prostatectomy as biomarkers for prostate cancer progression. They identified an HLA allele associated with disease recurrence in patients with low-intermediate risk prostate cancer and an allele associated with rapid disease recurrence in patients with high-risk prostate cancer.
Pedraz-Valdunciel et al customised a circRNA panel to detect circRNA expression both in NSCLC cells and formalin-fixed paraffin-embedded (FFPE) tissues and integrated machine learning approaches allowing discrimination of NSCLC from nontumor controls with an 8-circRNA signature and classified early-stage NSCLC samples using an additional 4-circRNA signature.
Adam et al developed a classification algorithm using machine learning to identify molecular subtypes of colorectal cancer based on microRNAs, given their role in tumor heterogeneity, instead of messenger RNAs and evaluated its potential use in clinical workflows.
3) Leveraging sophisticated bioinformatic algorithms and high-throughput screening platforms new layers of patient stratification have surfaced, facilitating patient monitoring and biomarker selection for post-treatment decisions.
Cani et al proposed a single-cell DNA sequencing pipeline for tracking circulating tumor cells from whole-blood samples of patients with advanced ER-POS/HER2-negative breast cancer and reported novel treatment avenues for 73% of patients by targeting PIK3CA, FGFR and KIT.
Mainguené et al retrospectively assessed HPV integration sites and signatures in HPV positive patients with head and neck squamous cell carcinoma (HNSCC) and identified HPV16 in 90% of the analyzed cohort confirming five previously described mechanistic signatures of HPV integration as well as recurrent targeting of several cancer genes such as PDL1 and MYC upon HPV integration.
Dziadziuszko et al evaluated the clinical validity of a non-invasive in vitro NGS platform to detect genomic alterations in plasma circulating tumour DNA (ctDNA) as an aid in identifying patients with NTRK-fp or ROS1-fp tumours and found that it can complement tissue-based testing for the identification of patients who may benefit from entrectinib treatment.
Ibrahim et al explored genome-wide methylation profiles of different cancer types and developed a three-step computational approach identifying tumor-specific CpG markers comprising a type-specific prediction model to differentiate between tumor types.
4) Finally, utilising the latest technological advancements new studies have been able to better characterise previously hidden aspects of disease or help elucidate new drug targets.
Giliberto et al established a drug testing pipeline to examine ex vivo drug responses in multiple myeloma which provided results within a clinically actionable 5-day time frame and identified synergistic drug efficacies in patient-derived MM cells that may aid future therapy choices.
Symonds et al assessed BCAT1 and IKZF1 methylation levels to quantify ctDNA and investigated the potential for identifying residual disease due to treatment failure.
Heitink et al described a pipeline for the identification of tumor suppressor genes that drive breast tumorigenesis via an in vivo genome-wide CRISPR/Cas9 screen highlighting mutagenesis of Trp53 and either Prkar1a, Axin1, or Pten markedly accelerated tumor development compared to Trp53-only mutants.
You may also access our collection of highlighted articles here:
Would your own research fit well within this collection? Then we encourage you to submit your manuscript to our journal.
Molecular Oncology is an Open Access international journal that highlights new discoveries, approaches, as well as technical developments, in basic, clinical and discovery-driven translational cancer research.




Join the FEBS Network today
Joining the FEBS Network’s molecular life sciences community enables you to access special content on the site, present your profile, 'follow' contributors, 'comment' on and 'like' content, post your own content, and set up a tailored email digest for updates.