Deep learning and genomics: predicting gene expression from DNA sequence

The human genome is enormously complex. Before the first draft of the genome was completed in 2003, there was an optimism that knowing its sequence could smoothly translate into cures and treatments for various diseases. Unfortunately, the scientific community quickly discovered that having the DNA sequence is still many steps away from understanding its emergent biological properties, such as the genetic basis of disease or high-level organismal traits. And certain sections of the genome are particularly mysterious – only about 2% of the roughly 3 billion DNA bases in the human genome correspond to genes that encode for proteins, meaning that 98% of the genome is composed of 'non-coding' regions of often unclear function. At DeepMind, one of our core missions is to use AI to help shed light on complex scientific domains such as genomics, with the aim of accelerating scientific progress and supporting future human health advances.
Understanding the principles of gene expression and its regulation is a grand challenge for basic research and human health. Gene expression refers to how many molecules of RNA are produced ('transcribed') from DNA. RNA is itself the precursor to proteins, which are the pervasive molecular workhorses of the cell. While every cell in the human body has a complete copy of that person’s DNA, not all genes are used or expressed in every cell type at all times. The mechanism known as gene regulation controls which genes are expressed, when, and in which cells or tissues. Errors in gene regulation are common causes of genetic disease – numerous disorders are caused by mutations that change the expression levels of genes. Ultimately, gene function and regulation are encoded within the linear DNA sequence. Accordingly, a long-standing problem in regulatory genomics is that of predicting gene expression purely from DNA sequence.
An overview of our gene expression model
In our recent Nature Methods paper titled Effective gene expression prediction from sequence by integrating long-range interactions, we – in collaboration with our Alphabet colleagues at Calico – developed a neural network model called Enformer that led to greatly increased accuracy in predicting gene expression from DNA sequence alone. We also show that Enformer more accurately predicts the effects of mutations on gene expression, and can be used to link regulatory regions to the genes they affect. To advance further study of gene regulation, we make our model’s predictions of the effects of common genetic variants openly available here. We have also open sourced our model code, so that other researchers can use our model to generate predictions on new sequences and variants, interpret mutations potentially involved in disease, or directly build on our architecture in their own work.
Previous deep learning work in gene expression prediction has typically used convolutional layers as its core architecture. Convolutional layers can be powerful motif extractors, but are limited in their ability to model the relationship between distant sections of the input sequence. The human genome is characterised by long-range interactions between different regions – for example, one of the most common DNA elements regulating gene expression in the human genome are 'enhancers', which can dramatically influence a gene’s expression levels from potentially millions of bases away. This means that a successful gene expression prediction model must both have visibility of a long input string of DNA and an ability to conceptually connect distant regions. Motivated by our collaborators’ previous modelling work in the Basenji2 paper and the knowledge that regulatory DNA elements can influence expression at long distances, we saw the need for a fundamental architectural change to capture long sequences.
In order to integrate a much wider DNA context, we developed a new model based on Transformers, a deep learning architecture common in natural language processing. Transformers excel at comprehending long passages of text, and accordingly, they are the foundation for current state-of-the-art models in machine translation, summarisation, and text generation. We adapted Transformers to 'read' vastly extended DNA sequences – by considering DNA sequences more than five times (i.e. 200,000 base pairs) the length of previous methods and using attention, which is the core algorithmic technique of Transformers, to capture element relationships. In this way, our architecture can better see and integrate the influence of enhancers and other regulatory regions on gene expression. Finally, in alignment with previous work in the field, we found that simultaneously predicting multiple different molecular phenomena and considering multiple species further improved human gene expression predictions, highlighting the core shared biology across different processes and organisms.
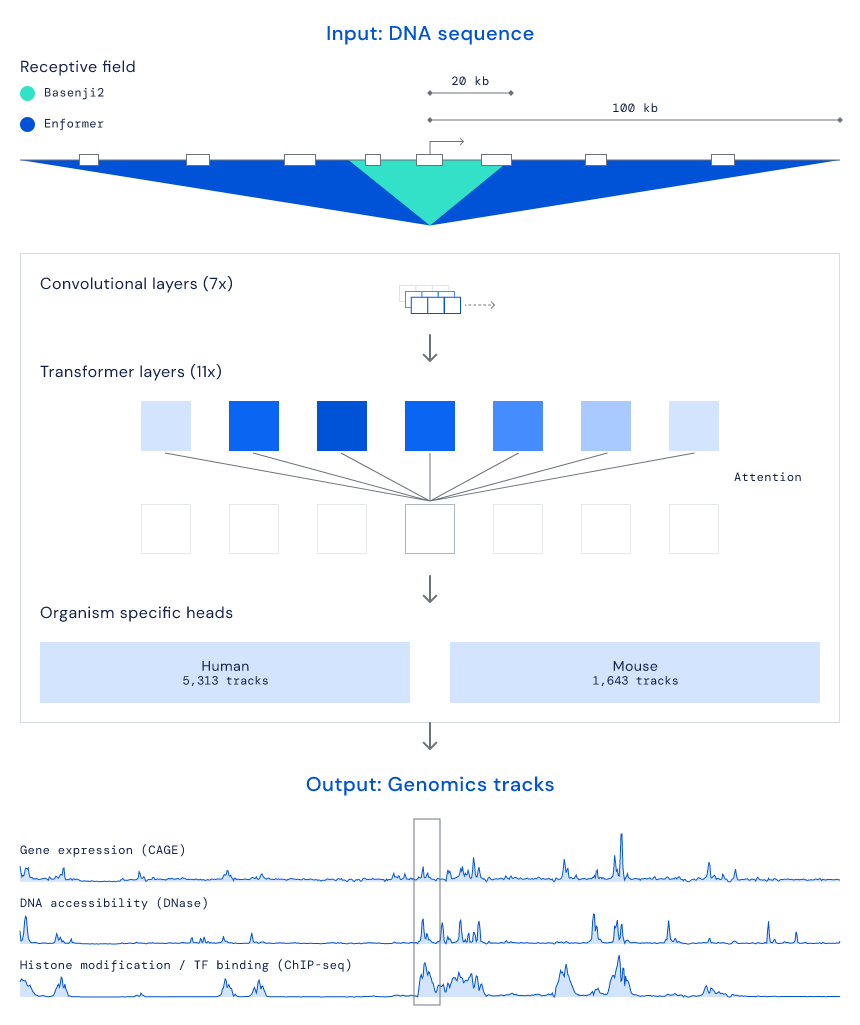
Interpreting and applying Enformer
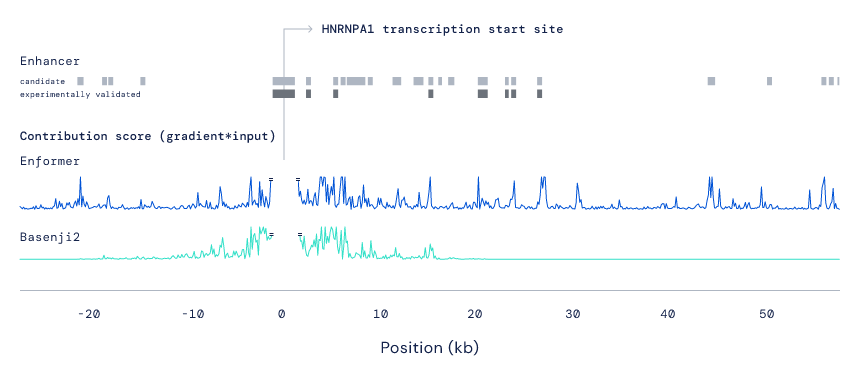
Although an improved black box gene expression model is useful, we also wanted to understand the internal mechanics of how Enformer interprets DNA sequences, and whether these align with our current understanding of the underlying biology. We analysed which regions of the DNA sequence were most crucial for the model’s predictions (using contribution scores) and explored the learnt relationships between sub-regions in the sequence (using attention weights). Matching the biological intuition, we observed that the model paid attention to enhancers even if these were located more than 50,000 base pairs away from the gene they affect. Predicting which enhancers affect the expression of which genes remains a major unsolved biological problem, so we were pleased to see that Enformer’s contribution scores perform comparably with existing state-of-the-art methods developed specifically for this task, despite never being directly trained for it. Enformer also learned to pay attention to another fundamental category of regulatory elements in the genome called 'insulators', which separate two independently regulated regions of DNA. These observations suggest that Enformer has learned some understanding of DNA sequences that is potentially transferable to other DNA-related problems.
The main application of our new model is predicting the effects of mutations on gene expression (where a mutation or 'variant' is a change to the DNA letters). Compared to previous models, Enformer is substantially more accurate at this task, both in the case of natural genetic variants in the general population and designed synthetic variants that alter important regulatory sequences. For example, Enformer outperforms all other entries in the CAGI5 challenge of predicting the effects of enhancer and promoter mutations on gene expression. Additionally, by relying solely on DNA sequence as input, Enformer has several advantages over other variant effect prediction methods: it does not rely on evolutionary conservation statistics, it can take any DNA sequence as input, and crucially it can predict whether a variant will increase or decrease a gene’s expression (rather than just predicting a change, but not specifying the directionality). To explore the utility of Enformer in dissecting the potential mode of action of a mutation, we examined the immune-related variant rs11644125, leading to a functional hypothesis related to disrupted transcription factor binding (see figure and caption below for the case study).
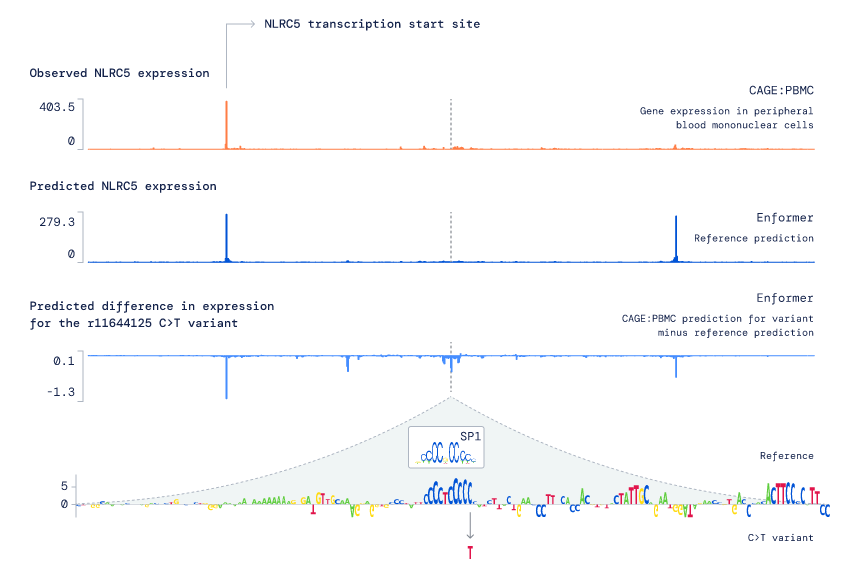
A case study of applying Enformer to understand the potential mechanism of action of a particular variant (rs11644125) associated with lower white blood cell counts. First, we observed that Enformer predicts lower NLRC5 expression in the presence of the variant (where NLRC5 is an immune response gene). By systematically mutating every position surrounding the variant (position shown as vertical dotted line) and predicting the resulting change on NLRC5 gene expression, we found that mutations in the vicinity of rs11644125 generally also lead to lower predicted expression of NLRC5. Further analysis of this region shows that these mutations change the known binding motif of a transcription factor called SP1, thereby suggesting that the mechanism underlying rs11644125’s effect on white blood cell counts is lower NLRC5 gene expression due to perturbed SP1 binding.
This enhanced ability of Enformer to understand the effects of variants will be useful for interpreting the growing number of disease-associated variants discovered by genome-wide association studies (GWAS). Indeed, the vast majority of variants associated with complex genetic diseases are predominantly located in the non-coding region of the genome, meaning that complex disease is likely more commonly associated with alterations to gene expression rather than direct changes to protein structure. Although GWA studies have uncovered a treasure trove of associations between low-level DNA sequence changes and high-level human traits, they often identify many spuriously-correlated variants alongside true causal variants because of the inherent genetic correlation among positions in the genome. In this new emerging era of deep learning in molecular biology, we believe that computational tools such as Enformer will be used to help distinguish true variant-trait associations from noisy false positives, thereby helping to uncover new causal mechanisms of disease.
Our understanding of the genome’s function is still very much in its early stages, but we see Enformer as a step forward towards modelling and deciphering the complexity of genomic sequences.
This post is a version of DeepMind's blog post Predicting gene expression with AI. The authors are Ziga Avsec, Staff Research Scientists, and Natasha Latysheva, Research Engineer, both at DeepMind.
Photo by chiranjeevi a on Unsplash





Join the FEBS Network today
Joining the FEBS Network’s molecular life sciences community enables you to access special content on the site, present your profile, 'follow' contributors, 'comment' on and 'like' content, post your own content, and set up a tailored email digest for updates.